ICS 87.040
G 51
代替 GB 18582—2001
IndOOr decorating and refurbishing materials— LinIit Of harmful SUbStanCeS Of interior architectural COatingS
2008-04-01 发布
中 华人民 共和国
国家标准
室内装饰装修材料
内墙涂料中有害物质限量
GB 18582—2008
*
中国标准出版社出版发行
北京复兴门外三里河北街16号
邮政编码= 100045
网址 WWW. spc. net. Cn
电话:68523946 68517548
中国标准出版社秦皇岛印刷厂印刷
各地新华书店经销
⅜
开本880X1230 1/16 印张 L 25 字数33千字
2008年6月第一版2008年6月第一次印刷
*
书号:155066 • 1-31710 ,
如有印装差错由本社发行中心调换 版权专有侵权必究
举报电话:(010)68533533
本标准代替GB 18582—2OO1≪室内装饰装修材料 内墙涂料中有害物质限量》。
本标准与GB 18582—2001相比主要技术差异:
——范围中增加了水性墙面腻子,并对其规定了有害物质限量值;
——水性墙面涂料中挥发性有机化合物的限量值大幅度降低,表示方法改为产品中除水后的挥发 性有机化合物的含量;
——游离甲醛计量单位改变,其限量值更加严格;
——增加了苯、甲苯、乙苯和二甲苯总和控制项目;
——增加了挥发性有机化合物的定义,测试方法由总挥发物扣除水分改为用气相色谱分析技术分 离被测样品中各种挥发性有机化合物并定性鉴定和定量分析;
——修改完善了游离甲醛和可溶性重金属的测试方法;
——建立了苯、甲苯、乙苯和二甲苯总和的测试方法,并将其与测试挥发性有机化合物方法相结合。
本标准的附录A、附录B、附录C、附录D为规范性附录。
本标准由中国石油和化学工业协会提出。
本标准由全国涂料和颜料标准化技术委员会(SAC/TC 5)归口。
本标准负责起草单位:中海油常州涂料化工研究院(国家涂料质量监督检验中心)、北京微量化学研 究所、上海市涂料研究所。
本标准参加起草单位:中国涂料工业协会、上海市建筑科学研究院、中国建筑科学研究院、立邦涂料 (中国)有限公司、广东华润涂料有限公司、广东嘉宝莉化工有限公司、卜内门太古漆油(中国)有限公司、 上海中南建筑材料公司、广东美涂士化工有限公司、莆田市三江化学工业有限公司、中华制漆(深圳)有 限公司、南宝树脂(中国)有限公司、江苏大象东亚制漆有限公司、罗门哈斯(中国)投资有限公司、杭州油 漆有限公司、南京天祥涂料有限公司、常州光辉化工有限公司、东莞大宝化工制品有限公司、上海富臣化 工有限公司、广东巴德士化工有限公司。
本标准主要起草人:张俊智、赵玲、冯世芳、黄宁、于滨、尹建武、张卫群、曹海华、黄添源、杨勇、 龚万森、段质美、寇辉、王代民、熊荣、王大期、李锋、彭冬华、凌萍、姜亚琴、杨少武、杨卫疆、姜方群、 徐凯斌、刘琳、黄建华、叶荣森、方学平。
本标准2001年12月10日首次发布,本次为第一次修订。
本标准委托全国涂料和颜料标准化技术委员会负责解释。
室内装饰装修材料
内墙涂料中有害物质限量
1范围
本标准规定了室内装饰装修用水性墙面涂料(包括面漆和底漆)和水性墙面腻子中对人体有害物质 容许限量的要求、试验方法、检验规则、包装标志、涂装安全及防护。
本标准适用于各类室内装饰装修用水性墙面涂料和水性墙面腻子。
2规范性引用文件
下列文件中的条款通过本标准的引用而成为本标准的条款。凡是注日期的引用文件,其随后所有 的修改单(不包括勘误的内容)或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究 是否可使用这些文件的最新版本。凡是不注日期的引用文件,其最新版本适用于本标准。
GB/T 601化学试剂标准滴定溶液的制备
GB/T 1250极限数值的表示方法和判定方法
GB/T 3186—2006色漆、清漆和色漆与清漆用原材料取样(ISo 15528 = 2000,IDT)
GB/T 6682 分析试验室用水规格和试验方法(GB/T 6682—1992,neq ISO 3696 = 1987)
GB/T 6750 色漆和清漆 密度的测定 比重瓶法(GB/T 6750—2007, ISO 2811-1 : 1997,IDT)
GB/T 9750涂料产品包装标志
3术语和定义
下列术语和定义适用于本标准。
3. 1
挥发性有机化合物(VOC) VOIatile OrganiC COmPOUndS
在101. 3 kPa标准压力下,任何初沸点低于或等于250°C的有机化合物。
3.2
挥发性有机化合物含量 volatile OrganiC COmPOUndS COntent
按规定的测试方法测试产品所得到的挥发性有机化合物的含量。
注1:墙面涂料为产品扣除水分后的挥发性有机化合物的含量,以克每升(g∕L)表示。
注2:墙面腻子为产品不扣除水分的挥发性有机化合物的含量,以克每千克(g/kg)表示。
4 要求
产品中有害物质限量应符合表1的要求。
表1有害物质限量的要求
|
项 目 |
限量值 | ||
|
水性墙面涂料a |
水性墙面腻子b | ||
|
挥发性有机化合物含量(Voe) |
≤ |
120 g/L |
15 g/kg |
|
苯、甲苯、乙苯、二甲苯总和/(mg/kg ) ≤ |
300 | ||
|
游离甲醛/(mg/kg) |
≤ |
100 | |
表1 (续)
|
项 目 |
限量值 | |
|
水性墙面涂料a 水性墙面腻子b | ||
|
可溶性重金属/(mg/kg) ≤ |
铅Pb |
90 |
|
镉Cd |
75 | |
|
馅Cr |
60 | |
|
汞Hg |
60 | |
|
a涂料产品所有项目均不考虑比。 b膏状腻子所有项目均不考虑稀释配比;粉状腻子除可溶性重金属项目直接测邸体外,其余3项按产品规定的 配比将粉体与水或胶黏剂.等其他液体混合后测试。如配比为某一范围时,应按照水用量最小、胶黏剂等其他液 体用量最大的配比混洽后测试。 | ||
5试验方法
5.1 取样 IGl 倉[ I
产品取样应按GB/T 3186 2006的规定证行
5.2试验方法
5. 2. 1挥发性有机化合物含S(VoC)的测试按附录A和附录B的规定进行,涂料产品测试结果的计 算按附录A中A. 7. 2进行。腻子产品测试结果的计算按附录ʌ ψ A. 7.1进行。
注:所有腻子样晶不做水分含量和密度的测试。
5.2.2苯、甲苯、乙苯和二甲苯总和的测试按附录A的规定进行。测试结果的计算按附录A中A.7.3
进行。 .(I: I I
5.2.3游离甲醛的测试按附录C的规定进行。
5.2.4可溶性重金属(铅、镉、铭和求)的测试按附录D的规定进行。粉状腻子苴接用粉体测试。
6检验规则
6. 1本标准所列的全部要求均为型式检验项目。
6. 1. 1在正常生产情况下,每年至少进行一次型式检验。
6.1.2有下列情况之一时应随时进行型式检验:
新产品最初定型时;
——产品异地生产时;
——生产配方、工艺及原材料有较大改变时;
——停产三个月后又恢复生产时。
6.2检验结果的判定
6. 2. 1检验结果的判定按GB/T 1250中修约值比较法进行。
6.2.2粉状腻子报出检验结果时应同时注明配制比例。
6. 2. 3所有项目的检验结果均达到本标准的要求时,产品为符合本标准要求。
7包装标志
产品包装标志除应符合GB/T 9750的规定外,按本标准检验合格的产品可在包装标志上明示。
8涂装安全及防护
8. 1涂装时应保证室内通风良好。
8.2涂装时施工人员应穿戴好必要的防护用品。
8. 3涂装完成后继续保持室内空气流通。
附录 A
(规范性附录)
挥发性有机化合物及苯、甲苯、乙苯和二甲苯总和含量的测试气相色谱法
A. 1范围
本方法规定了水性墙面涂料和水性墙面腻子中挥发性有机化介物(VCC及苯、甲苯、乙苯和二甲 苯总和含量的测试方法
本方法适用于VOf的含代大「或等「0.1%、且小于或等J'- lɔ/的涂料及儿原料的测试。
A.2原理
试样经稀释后顼Jl过气相色谱分析技术使样品中各种挥发件仃机化合物分离.是性鉴定被测化合物 后,用内标法测试其岔驼.
A.3材料和试剂
A.3.1载气:氮气.纯rr
A. 3.2 燃气:氢气,纯度-99.995%。
A. 3. 3助燃气:空气。
A. 3.4 辅助气体(隔垫吹扫和尾吹气):与我气只S相同性质的藐气。
A.3.5内标物:试样中不存在的化合物∙IL该化介物能够与色谱图上其他成分完全分离。纯度至少为 99%,或已知纯度「例如:异丁醇、乙二醇单丁醵、乙二褲二F卩•謎、二乙二醇二甲醍等。
A. 3. 6校准化合物
本标准中校准化合物包括甲醇、乙醇、正丙成、界丙F序、小:「醉、异丁醇、苯、甲苯、乙苯、二甲苯、三乙 胺、二甲基乙醇胺、2-氨基2甲基十丙醇、乙二醇、1,2-丙二醇、1,3-丙二醇、二乙二醇、乙二醇单丁醍、二 乙二醇单丁醵、二乙二醇乙醍醋酸酯、二乙.:酵丁謎船酸酯、2,2,,I-三甲基-1,3-戊二醇。纯度至少为 99%,或已知纯度。
A.3.7稀释溶剂:用于稀释试样的行机溶剂W、含有任何I:扰测试的物质。纯度至少为99%,或已知 纯度。例如:乙腊、甲醇或四氢味喃等溶剂。
A.3.8标记物:用于按VoC定义区分VOC组分与非VOC组分的化合物。本标准中为己二酸二乙酯 (沸点 251OC)O
A. 4仪器设备
A.4. 1气相色谱仪,具有以下配置:
A.4. 1. 1分流装置的进样口,并且汽化室内祠可更换。
A.4. 1.2程序升温控制器。
A.4. 1.3检测器:可以使用下列三种检测器中的任意一种:
A.4. 1.3. 1火焰离子化检测器(FlD)。
A. 4. 1. 3. 2已校准并调谐的质谱仪或其他质量选择检测器。
A. 4. 1.3.3已校准的傅立叶变换红外光谱仪(FT-IR光谱仪)O
注:如果选用A.4. 1.3.2或A.4. 1. 3. 3检测器对分离出的组分进行定性鉴定,仪器应与气相色谱仪相连并根据仪 器制造商的相关说明进行操作。
A. 4. 1.4色谱柱:聚二甲基硅氧烷毛细管柱或6%腊丙苯基/94%聚二甲基硅氧烷毛细管柱、聚乙二醇 毛细管柱。
4
A. 4. 2进样器:微量注射器,10 μLa
A.4. 3配样瓶:约20 mL的玻璃瓶,具有可密封的瓶盖。
A. 4. 4天平:精度0. 1 mg0
A.5气相色谱测试条件
A.5. 1色谱条件1
色谱柱(基本柱):聚二甲基硅氧烷毛细管柱,30 m×0.32 mm×1.0 Ym;
进样口温度:260°C;
检测器:FID,温度:280°C;
柱温:程序升温,45°C保持4 min,然后以8 °C∕min升至230°C保持10 min;
分流比:分流进样,分流比可调;
进样量:1. 0 μLo
A.5.2色谱条件2
色谱柱(基本柱):6%月青丙苯基/94%聚二甲基硅氧烷毛细管柱,60 m×0. 32 mm×1.0 Mm;
进样口温度:250°C;
检测器:FID,温度:260°C;
柱温:程序升温,80°C保持1 min,然后以10 °C∕min升至230°C保持15 min;
分流比:分流进样,分流比可调;
进样量:1. O μLo
A. 5. 3色谱条件3
色谱柱(确认柱):聚乙二醇毛细管柱,30 m×0.25 mm×0. 25 μm;
进样口温度:24OOC ;
检测器:FID,温度:250°C;
柱温:程序升温,60°C保持1 min,然后以20 °C∕min升至240°C保持20 min;
分流比:分流进样,分流比可调;
进样量:1. O μLo
注:也可根据所用气相色谱仪的性能及待测试样的实际情况选择最佳的气相色谱测试条件。
A.6测试步骤
A.6. 1密度
密度的测试按GB/T 6750进行。
A. 6. 2水分含量
水分含量的测试按附录B进行。
A.6.3挥发性有机化合物及苯、甲苯、乙苯和二甲苯总和含量
A.6. 3. 1色谱仪参数优化
按A. 5中的色谱条件,每次都应该使用已知的校准化合物对其进行最优化处理,使仪器的灵敏度、 稳定性和分离效果处于最佳状态。
A.6.3. 2 定性分析
定性鉴定试样中有无A. 3. 6中的校准化合物。优先选用的方法是气相色谱仪与质量选择检测器 (A. 4. 1. 3. 2)或FT-IR光谱仪(A. 4.1. 3. 3)联用,并使用A. 5中给岀的气相色谱测试条件。也可利用 气相色谱仪,釆用火焰离子化检测器(FlD)(A. 4.1. 3. 1)和A.4. 1.4中的色谱柱,并使用A. 5中给出的 气相色谱测试条件,分别记录A. 3. 6中校准化合物在两根色谱柱(所选择的两根柱子的极性差别应尽 可能大,例如6%月青丙苯基/94%聚二甲基硅氧烷毛细管柱和聚乙二醇毛细管柱)上的色谱图;在相同的 色谱测试条件下,对被测试样做出色谱图后对比定性。
A. 6. 3. 3 校准
A. 6. 3. 3. 1校准样品的配制:分别称取一定量(精确至O. 1 mg)A. 6. 3. 2鉴定出的各种校准化合物于 配样瓶(A.4.3)中,称取的质量与待测试样中各自的含量应在同一数量级;再称取与待测化合物相同数 量级的内标物(A. 3. 5)于同一配样瓶中,用稀释溶剂(A. 3. 7)稀释混合物,密封配样瓶并摇匀。
A. 6. 3. 3. 2相对校正因子的测试:在与测试试样相同的色谱测试条件下按A. 6. 3. 1的规定优化仪器 参数。将适当数量的校准化合物注入气相色谱仪中,记录色谱图。按式(A.1)分别计算每种化合物的 相对校正因子:
式中:
Ri--化合物i的相对校正因子;
mCi--校准混合物中化合物i的质量,单位为克(g);
mis——校准混合物中内标物的质量,单位为克(g);
AS--内标物的峰面积;
Ac-——化合物/的峰面积。
Ri值取两次测试结果的平均值,其相对偏差应小于5%,保留3位有效数字。
A. 6. 3. 3. 3若出现A. 3. 6中校准化合物之外的未知化合物色谱峰,则假设其相对于异丁醇的校正因 子为1.0。
A. 6. 3. 4试样的测试
A. 6. 3. 4. 1试样的配制:称取搅拌均匀后的试样1 g(精确至0.1 mg)以及与被测物质量近似相等的内 标物(A. 3. 5)于配样瓶(A. 4. 3)中,加入10 mL稀释溶剂(A. 3. 7)稀释试样,密封配样瓶并摇匀。
A.6.3.4.2按校准时的最优化条件设定仪器参数。
A.6.3.4.3将标记物(A. 3. 8)注入气相色谱仪中,记录其在聚二甲基硅氧烷毛细管柱或6%腊丙苯 基/94%聚二甲基硅氧烷毛细管柱上的保留时间,以便按3. 1给出的VoC定义确定色谱图中的积分 终点。
A. 6.3.4.4将IfXL按A. 6. 3.4. 1配制的试样注入气相色谱仪中,记录色谱图并记录各种保留时间低 于标记物的化合物峰面积(除稀释溶剂外),然后按式(A. 2)分别计算试样中所含的各种化合物的质量 分数。
式中:
WI——测试试样中被测化合物i的质量分数,单位为克每克(g/g);
Rl——被测化合物i的相对校正因子;
mis 内标物的质量,单位为克(g);
ms——测试试样的质量,单位为克(g);
AS 内标物的峰面积;
Ai——被测化合物i的峰面积。
平行测试两次,叫值取两次测试结果的平均值。
A. 7计算
A.7. 1腻子产品按式(A. 3)计算VoC含量: W(VOC) = ɪɔwɪ. X 1 000 ..............................( A. 3 )
式中:
W(VOC)——腻子产品的VOC含量,单位为克每千克(g∕kg);
Wf——测试试样中被测化合物i的质量分数,单位为克每克(g/g);
1 OOO——转换因子。
测试方法检出限:1 g∕kgo
A. 7. 2 涂料产品按式(A. 4)计算VoC含量:
Vw
jo(VOC) = -~XP × 1 000 ..............................( A.4 )
WW
1 — P ∕μ∖---
S QW
式中:
Q(VOC)——涂料产品的VoC含量,单位为克每升(g∕L);
wi.——测试试样中被测化合物i的质量分数,单位为克每克(g∕g);
WW——测试试样中水的质量分数,单位为克每克(g/g);
PS--试样的密度,单位为克每毫升(g∕mL);
PW--水的密度,单位为克每毫升(g∕mL);
1 000——转换因子。
测试方法检出限:2 g∕Lo
A.7.3涂料和腻子产品中苯、甲苯、乙苯和二甲苯总和的计算
A. 7. 3. 1先按式(A. 2)分别计算苯、甲苯、乙苯和二甲苯各自的质量分数叫,然后按式(A. 5)计算产品 中苯、甲苯、乙苯和二甲苯含量的总和:
Wb = ɪɔw. × IO6 ..............................( A. 5 )
式中:
Wb——产品中苯、甲苯、乙苯和二甲苯总和的含量,单位为毫克每千克(mg/kg);
Wi——测试试样中被测组分z(苯、甲苯、乙苯和二甲苯)的质量分数,单位为克每克(g∕g);
IO6——转换因子。
A. 7. 3. 2测试方法检出限:4种苯系物总和50 mg/kgO
A. 8精密度
A. 8. 1 重复性
同一操作者两次测试结果的相对偏差小于10%。
A. 8. 2再现性
不同实验室间测试结果的相对偏差小于20%。
附录B (规范性附录) 水分含量的测试
本标准中的水分含量釆用气相色谱法或卡尔。费休法测试。气相色谱法为仲裁方法。
B. 1气相色谱法
B. 1. 1试剂和材料
B. 1. 1. 1蒸馆水:符合GB/T 6682中三级水的要求。
B. 1. 1.2稀释溶剂:无水二甲基甲酰胺(DMF),分析纯。
B. 1. 1.3内标物:无水异丙醇,分析纯。
B. 1. 1.4载气:氢气或氮气,纯度不小于99.995⅜o
B. 1.2仪器设备
B. 1.2. 1气相色谱仪:配有热导检测器及程序升温控制器。
B. 1.2.2色谱柱:填装高分子多孔微球的不锈钢柱。
B. 1.2.3进样器:微量注射器,10 μLo
B. 1.2.4配样瓶:约IomL的玻璃瓶,具有可密封的瓶盖。
B. 1.2.5 天平:精度 0.1 mgo
B. 1.3气相色谱测试条件
色谱柱:柱长1 m,外径3. 2 mm,填装177 Vm~250 μm高分子多孔微球的不锈钢柱。
汽化室温度:20OOCo
检测器:温度240OC ,电流150 mA。
柱温:对于程序升温,80°C保持5 min,然后以30 °C∕min升至170°C保持5 min;对于恒温,柱温为 90°C,在异丙醇完全流出后,将柱温升至170oC,待DMF出完。若继续测试,再把柱温降到90oCO
注:也可根据所用气相色谱仪的性能及待测试样的实际情况选择最佳的气相色谱测试条件。
B. 1.4测试步骤
B. 1.4. 1测试水的相对校正因子R
在同一配样瓶(B. 1. 2.4)中称取0. 2 g左右的蒸馅水(B. 1. 1. 1)和0. 2 g左右的异丙醇(B. L 1. 3), 精确至0. 1 mg,再加入2 mL的二甲基甲酰胺(B. 1. 1. 2),密封配样瓶并摇匀。用微量注射器 (B.1.2.3)吸取IVL配样瓶中的混合液注入色谱仪中,记录色谱图。按式(B. 1)计算水的相对校正因 子R:
式中:
R--水的相对校正因子;
mi——异丙醇质量,单位为克(g);
mw——水的质量,单位为克(g);
Ai——异丙醇的峰面积;
AW——水的峰面积。
若异丙醇和二甲基甲酰胺不是无水试剂,则以同样量的异丙醇和二甲基甲酰胺(混合液),但不加水 作为空白样,记录空白样中水的峰面积厶0。按式(B. 2)计算水的相对校正因子R:
8
Tni X (AW -~ ∙Ao) TnVI × Ai
√ B.2 )
式中:
R--水的相对校正因子;
mi——异丙醇质量,单位为克(g);
mvf--水的质量,单位为克(g);
Ai——异丙醇的峰面积;
-AW 水的峰面积;
AO--空白样中水的峰面积。
R值取两次测试结果的平均值,其相对偏差应小于5%,保留3位有效数字。
B. 1.4.2样品分析
称取搅拌均匀后的试样。.6 g以及与水含量近似相等的异丙醇(B. 1. 1. 3)于配样瓶(B. 1. 2. 4)中, 精确至0. 1 mg,再加入2.mL:二甲基甲酰胺(B. 1. 1. 2),密封配样瓶苦摇匀。、同时准备一个不加试样的 异丙醇和二甲基甲酰胺混合液作为斜'{样。用力採动渋祐认样的HZ样瓶15 min,放置5 min,使其沉淀 (为使试样尽快沉海,可在装有试样的配样瓶内加入儿粒小坡璃垸,然后用力摇动;也可使用低速离心机 使其沉淀)。用微量注射器(B. 1.2.3)吸取1 /丄配样瓶中的[力清液,注入色曲仪屮,记录色谱图。按 式(B. 3)计算试样中的水分含量:
硏,X 100 ..............................( B.3 )
IIl √x AtxR
WW一■试样中的水分含量的质诚分薮X ;
R——水的相对校正因子;
mi 异丙.醇质量,单位为克(R):
mS--试样的质量,单位为克(R);
Ai——异丙醺的峰面积;
AW 试样中水的峰面积;
AO—— 空白样中水的峰面弔:
平行测试两次.,取两次测试结果的平均值-保洛3位冇效数字,
B. 1.5精密度
B. 1.5. 1 重复性 〜一
同一操作者两次测试结果的相对偏差小于1. 6%。
B. 1.5.2再现性
不同实验室间测试结果的相对偏差小于5%。
B. 2卡尔.费休法
B.2. 1仪器设备
B.2. 1. 1卡尔。费休水分滴定仪。
B.2. 1.2 天平:精度 0.1 mg,l mgo
B. 2. 1.3微量注射器:10 μLo
B. 2. 1.4 滴瓶:30 mLo
B. 2. 1.5磁力搅拌器。
B.2. 1.6 烧杯:100 mLo
B.2. 1.7 培养皿。
B. 2.2 试剂
B.2.2. 1蒸德水:符合GB/T 6682中三级水的要求。
B.2.2.2卡尔≡费休试剂:选用合适的试剂(对于不含醛酮化合物的试样,试剂主要成分为碘、二氧化 硫、甲醇、有机碱。对于含有醛酮化合物的试样,应使用醛酮专用试剂,试剂主要成分为碘、咪喳、二氧化 硫、2-甲氧基乙醇、2-氯乙醇和三氯甲烷)。
B.2. 3 实验步骤
B. 2. 3. 1卡尔。费休滴定剂浓度的标定
在滴定仪(B. 2. 1.1)的滴定杯中加入新鲜卡尔。费休溶剂(B. 2. 2. 2)至液面覆盖电极端头,以 卡尔。费休滴定剂滴定至终点(漂移值VIo μg∕min)o用微量注射器(B. 2. 1. 3)将10 NL蒸馆水 (B. 2. 2. 1)注入滴定杯中,釆用减量法称得水的质量(精确至0. 1 mg),并将该质量输入到滴定仪中,用 卡尔。费休滴定剂滴定至终点,记录仪器显示的标定结果。
进行重复标定,直至相邻两次的标定值相差小于0. 01 mg∕mL,求出两次标定的平均值,将标定结 果输入到滴定仪中。
当检测环境的相对湿度小于70%时,应每周标定一次;相对湿度大于70%时,应每周标定两次;必 要时,随时标定。
B.2.3.2样品处理
若待测样品黏度较大,在卡尔。费休溶剂中不能很好分散,则需要将样品进行适量稀释。在烧杯 (B.2.1.6)中称取经搅拌均匀后的样品20 g(精确至1 mg),然后向烧杯内加入约20%的蒸馆水 (B. 2. 2. 1),准确记录称样量及加水量。将烧杯盖上培养皿(B. 2.1. 7),在磁力搅拌器(B. 2. 1. 5)上搅拌 10 min〜15 minO然后将稀释样品倒入滴瓶(B. 2. 1. 4)中备用。
注:对于在卡尔•费休溶剂中能很好分散的样品,可直接测试样品中的水分含量。对于加水20%后,在卡尔•费 休溶剂中仍不能很好分散的样品,可逐步增加稀释水量。
B.2.3.3水分含量的测定
在滴定仪(B. 2. 1. 1)的滴定杯中加入新鲜卡尔。费休溶剂(B. 2. 2. 2)至液面覆盖电极端头,以 卡尔。费休滴定剂滴定至终点。向滴定杯中加入1滴按B. 2. 3. 2处理后的样品,釆用减量法称得加入 的样品质量(精确至0. 1 mg),并将该样品质量输入到滴定仪(B. 2. 1. 1)中。用卡尔。费休滴定剂滴定 至终点,记录仪器显示的测试结果。
平行测试两次,测试结果取平均值。两次测试结果的相对偏差小于1.5⅜o
测试3〜6次后应及时更换滴定杯中的卡尔O费休溶剂。
B.2.3.4 数据处理
样品经稀释处理后测得的水分含量按式(B. 4)计算:
式中:
WW——样品中实际水分含量的质量分数,% ;
w;——稀释样品测得的水分含量的质量分数平均值,%;
mS——稀释时所称样品的质量,单位为克(g);
mw——稀释时所加水的质量,单位为克(g)。
计算结果保留3位有效数字。
附录C (规范性附录) 游离甲醛含量的测试
C. 1原理
釆用蒸馆的方法将样品中的游离甲醛蒸出。在PH-6的乙酸-乙酸铉缓冲溶液中,德分中的甲醛 与乙酰丙酮在加热的条件下反应生成稳定的黄色络合物,冷却后在波长412 nm处进行吸光度测试。 根据标准工作曲线,计算试样中游离甲醛的含量
C.2试剂
分析测试中仅采用已确认为分析纯的试剂•所川水符合GB 丁 6储?屮 耘水的要求。所用溶液除 另有说明外,均应按羸GB/T 601中的耍求逬行配制,
C.2. 1 乙酸铉。IlS / ⅜ ⅜
C.2.2 冰乙酸:(O=LO55 g/mL
C. 2. 3 乙酰丙煎:侦.975 g∕mLo
C.2.4乙酰丙酮溶液:体积分数为().2尹/.祢収25 g乙駿铉(C∙2. I).丿川诂債水溶解,加3mL冰乙酸 (C. 2. 2)和0. 25 mL已蒸储过的乙酰丙闹试剂(C. 2. 3),移入100 mL容量瓶中,用水稀释至刻度,调整 pH = 6o此溶液于2°C〜5°C贮冇,可稳定•个月
C.2.5 碘溶液:c(l/2IJ-0. 1 HIOl∕I,.
C. 2. 6氢氧化钠溶液:1 mol∕Lo
C. 2. 7 盐酸溶液:1 mol∕LO
C. 2. 8硫代硫酸钠标准溶液:c(\H&(),)- 0. 1 nɪol L.并按照GBT' 6<)I拔行标定。
C.2.9淀粉溶液fl-g∕100 mL,称取.Ig淀粉.川少M水调成糊状,倒人1颯mL沸水中,呈透明溶液, 临用时配制。/ !
C.2. 10甲醛溶液:质量分数约为37 Z-
C.2. 11甲醛标准溶液:1 mg∕mL,移収:Z.S ∏ιL ∣∣儷溶液(("• IG). R「T (XemL容量瓶中,用水稀 释至刻度。
C.2. 12甲醛标准溶液的标定:移取20 mL待标定的甲醛标准溶滚((‘.2. 1 1.)「孩量瓶中,准确加入 25 mL碘溶液(C.2.5),再加入10 mL氢氧化钠溶液(C. 2.6).揺匀.「•带处•淨労5 mln后,加11 mL盐 酸溶液(C. 2. 7),用硫代硫酸钠标准溶液(C. 2. 8)滴定至淡黄色,加1 mL淀粉溶液(C. 2. 9),继续滴定 至蓝色刚刚消失为终点,记录所耗硫代硫.酸钠标准溶液体积V2(mL) O同时做空白样,记录所耗硫代硫 酸钠标准溶液体积Vι(mL)O按式(C.1)计算甲醛标准溶液的质量浓度。
ZHPwnλ (VI -v2) Xc(Na2S2O3) X 15 ZPl ʌ
(O(HCH(J) = ------------—............ ( C. 1 J
式中:
io(HCHO)—— 甲醛标准溶液的质量浓度,单位为毫克每毫升(mg/mL);
Vl——空白样滴定所耗的硫代硫酸钠标准溶液体积,单位为毫升(mL);
V2——甲醛溶液标定所耗的硫代硫酸钠标准溶液体积,单位为毫升(mL);
C(Na2S2O3)——硫代硫酸钠标准溶液的浓度,单位为摩尔每升(mol∕L);
15--甲醛摩尔质量的1/2;
20——标定时所移取的甲醛标准溶液体积,单位为毫升(mL)o
C. 2. 13甲醛标准稀释液:1O μg∕mL,移取10 mL按C. 2. 12标定过的甲醛标准溶液(C. 2. 11),置于 1 000 mL容量瓶中,用水稀释至刻度。
C.3仪器与设备
C.3. 1蒸馋装置:100 mL蒸馅瓶、蛇型冷凝管、馆分接受器。
C.3.2具塞刻度管:50mL(与C. 3. 1中馆分接受器为同一容器)。
C. 3. 3 移液管:1 mL、5 InLJo mL、20 mL、25 mLo
C. 3. 4加热设备:电加热套、水浴锅。
C.3.5天平:精度1 mgo
C.3.6紫外可见分光光度计。
C.4试验步骤
C.4. 1标准工作曲线的绘制
取数支具塞刻度管(C. 3. 2),分别移入 O. OO mL、0. 20 mL、0. 50 InL.1. OO mL、3. OO mL、5. OO mL、 8. OO mL甲醛标准稀释液(C. 2. 13),加水稀释至刻度,加入2.5 mL乙酰丙酮溶液(C. 2.4),摇匀。在 60°C恒温水浴中加热30 min,取出后冷却至室温,用10 mm比色皿(以水为参比)在紫外可见分光光度 计(C. 3. 6)上于412 nm波长处测试吸光度。
以具塞刻度管中的甲醛质量(Rg)为横坐标,相应的吸光度为纵坐标,绘制标准工作曲线。
C.4.2游离甲醛含量的测试
称取搅拌均匀后的试样2 g(精确至1 mg),置于50 mL的容量瓶中,加水摇匀,稀释至刻度。再用 移液管移取10 mL容量瓶中的试样水溶液,置于已预先加入10 mL水的蒸馅瓶(C. 3.1)中,在德分接受 器(C.3.2)中预先加入适量的水,浸没储分出口,馅分接收器的外部用冰水浴冷却,蒸馋装置见图C. Io 加热蒸储,使试样蒸至近干,取下馆分接收器,用水稀释至刻度,待测。
注:若待测试样在水中不易分散,则直接称取搅拌均匀后的试样0.4 g(精确至1 mg),置于已预先加入20 mL水的 蒸馋瓶中,轻轻摇匀,再进行蒸儲过程操作。
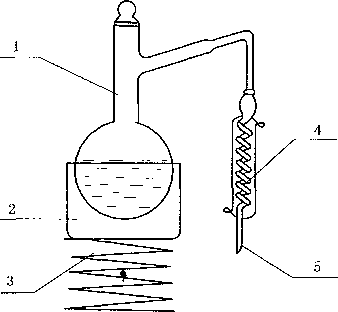
1— 蒸悔瓶;
2— —加热装置;
3— —升降台;
4--冷凝管;
5——连接接受装置。
图C. 1蒸馄装置示意图
在已定容的馆分接收器中加入2. 5 mL乙酰丙酮溶液(C. 2. 4),摇匀。在60°C恒温水浴中加热 30 min,取出后冷却至室温,用10 mm比色皿(以水为参比)在紫外可见分光光度计(C. 3. 6)上于 412 nm波长处测试吸光度。同时在相同条件下做空白样(水),测得空白样的吸光度。
将试样的吸光度减去空白样的吸光度,在标准工作曲线上查得相应的甲醛质量。
如果试验溶液中甲醛含量超过标准曲线最高点,需重新蒸偕试样,并适当稀释后再进行测试。
C.5结果的计算
C.5. 1游离甲醛含量按式(C.2)计算:
W = ^f ..............................( C.2 )
m
式中:
W——游离甲醛含量,单位为毫克每千克(mg/kg);
m——从标准工作曲线上查得的甲醛质量,单位为微克(昭);
m——样品质量,单位为克(g);
/——稀释因子。
C. 5. 2测试方法检出限:5 mg/kg。
C. 6 精密度
C. 6. 1重复性
当测试结果不大于100 mg/kg时,同一操作者两次测试结果的差值不大于10 mg/kg;当测试结果 大于100 mg/kg时,同一操作者两次测试结果的相对偏差不大于5%。
C. 6. 2 再现性
当测试结果不大于100 mg/kg时,不同试验室间测试结果的差值不大于20 mg/kg;当测试结果大 于100 mg/kg时,不同试验室间测试结果的相对偏差不大于10%。
附录D
(规范性附录)
可溶性铅、镉、铭、汞元素含量的测试
D. 1原理
用0. 07 mol/L盐酸溶液处理制成的涂料干膜,用火焰原子吸收光谱法测试试验溶液中可溶性铅、 镉、铭元素的含量,用氢化物发生原子吸收光谱法测试试验溶液中可溶性汞元素的含量。
D. 2试剂
分析测试中仅使用确认为分析纯的试剂,所用水符合GB/T 6682中三级水的要求。
D.2. 1 盐酸溶液:0.07 mol/Lo
D. 2.2盐酸:质量分数约为37%,密度约为1. 18 g∕cm3 O
D.2.3硝酸溶液:1 : 1(体积比)。
D.2.4 铅、镉、铭、汞标准溶液:浓度为100 mg∕L或1 OOo mg∕Lo
D.3仪器
D. 3. 1火焰原子吸收光谱仪:配备铅、镉、铭空心阴极灯,并装有可通入空气和乙焕的燃烧器。仪器工 作条件见表D.lo
D.3.2氢化物发生原子吸收光谱仪:配备汞空心阴极灯,并能与氢化物发生器配套使用。仪器工作条 件见表D. 1。
表D. 1火焰原子吸收光谱仪和氢化物发生原子吸收光谱仪工作条件
|
元素 |
测试波长/nm |
原子化方法 |
背景校正 |
|
铅(Pb) |
283. 3 |
空气-乙焕火焰法 |
笊灯 |
|
镉(Cd) |
228. 8 |
空气-乙焕火焰法 |
笊灯 |
|
铭(Cr) |
357. 9 |
空气-乙焕火焰法 |
笊灯 |
|
汞(Hg) |
253. 7 |
氢化物法 |
一 |
|
注:实验室可根据所用仪器的性能选择合适的工作参数(如灯电流、狭缝宽度、空气-乙焕比例、还原剂品种等), 使仪器处于最佳测试状况。 | |||
D. 3. 3 粉碎设备:粉碎机,剪刀等。
D. 3.4 不锈钢金属筛:孔径0.5 mmo
D.3.5天平:精度0. 1 mgo
D. 3. 6搅拌器:搅拌子外层应为聚四氟乙烯或玻璃[需用硝酸溶液(D. 2. 3)浸泡24 h,然后用水清洗并 干燥丄
D.3.7酸度计:精度为士0.2 PH单位。
D. 3. 8微孔滤膜:孔径0. 45 Vmo
D. 3.9 容量瓶:25 mL、50 mL、l。OmL。
D. 3. 10 移液管:1 InL>2 mL、5 mL、10 mL、25 mL。
D.3. 11系列化学容器:总容量为盐酸溶液提取剂体积的1.6-5. 0倍[需用硝酸溶液(D. 2. 3)浸泡
24 h,然后用水清洗并干燥丄
14
D. 4试验步骤
D.4. 1涂膜的制备
将待测样品搅拌均匀。按涂料产品规定的比例(稀释剂无须加入)混合各组分样品,搅拌均匀后,在 玻璃板或聚四氟乙烯板[需用硝酸溶液(D. 2. 3)浸泡24 h,然后用水清洗并干燥]上制备厚度适宜的涂 膜。待完全干燥[自干漆若烘干,温度不得超过(60 + 2)oC]后,取下涂膜,在室温下用粉碎设备(D.3.3) 将其粉碎,并用不锈钢金属筛(D. 3. 4)过筛后待处理。
注1:对不能被粉碎的涂膜(如弹性或塑性涂膜),可用干净的剪刀(D.3.3)将涂膜尽可能剪碎,无须过筛直接进行 样品处理。
注2:粉末状样品,直接进行样品处理。
D. 4.2样品处理
对制备的试样进行两次平行测试。
称取粉碎、过筛后的试样0.5 g(精确至0. 1 mg)置于化学容器(D. 3. 11)中,用移液管(D. 3. 10)加 入25 mL盐酸溶液(D. 2. l)o在搅拌器(D. 3. 6)上搅拌1 min后,用酸度计(D. 3.7)测其酸度。如果 PH值>1.5,用盐酸(D. 2. 2)调节PH值在1.0-1. 5之间。再在室温下连续搅拌1 h,然后放置1 ho 接着立即用微孔滤膜(D.3.8)过滤。过滤后的滤液应避光保存并应在一天内完成元素分析测试。若滤 液在进行元素分析测试前的保存时间超过1 d,应用盐酸(D.2.2)加以稳定,使保存的溶液浓度C(HCl) 约为 1 mol∕Lo
注1:如改变试样的称样量,则加入的盐酸溶液(D.2.1)体积应调整为试样量的50倍。
注2:在整个提取期间,应调节搅拌器的速度,以保持试样始终处于悬浮状态,同时应尽量避免溅出。
D.4.3标准参比溶液的配制
选用合适的容量瓶(D. 3. 9)和移液管(D. 3.10),用盐酸溶液(D. 2. 1)逐级稀释铅、镉、铭、汞标准溶 液(D.2.4),配制下列系列标准参比溶液(也可根据仪器及测试样品的情况确定标准参比溶液的浓度范 围):
铅(mg∕L) :0.0,2.5,5.0,10. 0,20. 0,30. 0 ;
镉(mg∕L) :0. 0,0. 1,0. 2,0. 5,1. 0;
铭(mg∕L) :0.0,l. 0,2. O,3.O,5.O;
汞(μg∕L)zO. 0,10. 0,20. 0,30. 0,40. Oo
注:系列标准参比溶液应在使用的当天配制。
D.4.4 测试
用火焰原子吸收光谱仪(D. 3. 1)及氢化物发生原子吸收光谱仪(D. 3. 2)分别测试标准参比溶液的 吸光度,仪器会以吸光度值对应浓度自动绘制出工作曲线。
同时测试试验溶液的吸光度。根据工作曲线和试验溶液的吸光度,仪器自动给出试验溶液中待测 元素的浓度值。如果试验溶液中被测元素的浓度超出工作曲线最高点,则应对试验溶液用盐酸溶液 (D. 2. 1)进行适当稀释后再测试。
如果两次测试结果(浓度值)的相对偏差大于10%。需按D.4试验步骤重做。
D.5结果的计算
D.5. 1试样中可溶性铅、镉、铭、汞元素的含量,按式(D.1)计算:
(P-PQW×F
W = ............. ・•・•,( IJ.丄)
m
式中:
W——试样中可溶性铅、镉、铭、汞元素的含量,单位为毫克每千克(mg/kg);
PQ——空白溶液(D. 2. 1)的测试浓度,单位为毫克每升(mg∕L);
P——试验溶液的测试浓度,单位为毫克每升(mg∕L);
V——盐酸溶液(D. 2. 1)的定容体积,单位为毫升(mL);
F——试验溶液的稀释倍数;
τn--称取的试样量,单位为克(g) o
D. 5.2 结果的校正
由于本测试方法精确度的原因,在测试结果的基础上需经校正得出最终的分析结果。即式(D. 1) 中的计算结果应减去该结果乘以表D. 2中相应元素的分析校正系数的值,作为该元素最终的分析结果 报出。
示例:铅的计算结果为120 mg∕kg,表D. 2中铅的分析校正系数为30%,则最终分析结果= 120 — 120X 30% = 84 mg/kgO
表D.2各元素分析校正系数
|
元素 |
铅(Pb) |
镉(Cd) |
铭(Cr) |
汞(Hg) |
|
分析校正系数/% |
30 |
30 |
30 |
50 |
D.6测试方法的检出限
按上述分析方法测试可溶性铅、镉、铭、汞元素含量,其检出限不应大于该元素限量(见表1 )的十分 之一。分析测试方法的检出限一般被认为是空白样测试值标准偏差的3倍,上述空白样测试值由实验 室测试。
D.7精密度
D. 7. 1 重复性
同一操作者两次测试结果的相对偏差小于20%。
D. 7. 2 再现性
不同试验室间测试结果的相对偏差小于33%。
版权专有侵权必究 书号:155066 • 1-31710
GB 18582-2008